

L’image d’un enfant souffrant m’étreint le cœur, mais elle irradie également un message d’espoir et de résilience infaillible. Elle capture l’essence de la persévérance et du courage face à l’adversité. Elle me rappelle que même dans les ténèbres les plus profondes, l’espoir scintille toujours, comme une flamme indomptable.
Introduction
Le sarcome d’Ewing est une tumeur qui se forme dans les os ou les tissus mous. Ce type de cancer survient principalement chez les enfants et les jeunes adultes, apparaissant souvent pendant l’adolescence. Les sarcomes d’Ewing sont les deuxièmes tumeurs osseuses primitives malignes les plus fréquentes de l’enfance après l’ostéosarcome. Ils proviennent généralement de la cavité médullaire avec l’invasion du système Haversien. Ce sarcome a été décrit pour la première fois par James Ewing, un pionnier dans le domaine de la recherche sur le cancer, en 1921.
Le sarcome d’Ewing affecte principalement les os longs, notamment le fémur, le tibia et l’humérus. Il peut se propager (métastaser) à d’autres parties du corps. Bien que relativement rare, environ 1,7 enfants de moins de 15 ans seront atteints de ce cancer.

Le sarcome d’Ewing peut se propager (métastaser) à d’autres parties du corps. Ce cancer est relativement rare. Environ 1.7 enfants 1 million en bas de 15 ans auront ce cancer.
Comprendre le Sarcome d’Ewing : Comment il se forme
Le sarcome d’Ewing est un cancer qui se développe dans les os ou les tissus mous, principalement chez les jeunes. Voici comment il se forme :
La Clé : Anomalie Génétique
- Mélange Chromosomique : Le sarcome d’Ewing commence par un problème génétique appelé translocation. Cela signifie que des parties des chromosomes 11 et 22 se mélangent de manière incorrecte, créant un nouveau gène bizarre appelé EWSR1-FLI1.
- Effets de ce Nouveau Gène : Ce gène bizarre produit une protéine anormale. Cette protéine perturbe la croissance normale des cellules osseuses, les empêchant de se différencier correctement en cellules spécialisées. Cela entraîne une croissance cellulaire hors de contrôle, caractéristique du cancer.
- Impact sur le Corps : La protéine anormale active des signaux dans les cellules qui favorisent la croissance rapide des tumeurs. Elle encourage également la formation de nouveaux vaisseaux sanguins autour de la tumeur, assurant ainsi son alimentation en nutriments et en oxygène.
En bref, le sarcome d’Ewing est provoqué par un problème génétique qui perturbe la croissance normale des os, conduisant à la formation d’une tumeur maligne.
Causes du sarcome d’Ewing
Le sarcome d’Ewing est une tumeur osseuse maligne rare qui affecte principalement les enfants, les adolescents et les jeunes adultes. Bien que ses causes exactes ne soient pas totalement élucidées, des facteurs génétiques et biologiques spécifiques jouent un rôle clé dans son développement. Voici un aperçu des principaux éléments associés à cette maladie.
Altérations génétiques spécifiques
La cause principale identifiée du sarcome d’Ewing est une translocation chromosomique spécifique, généralement entre les chromosomes 11 et 22. Cette anomalie génétique entraîne la fusion des gènes EWSR1 (sur le chromosome 22) et FLI1 (sur le chromosome 11). Le gène de fusion ainsi créé produit une protéine anormale qui dérégule l’expression génétique, favorisant ainsi la prolifération cellulaire incontrôlée. Ce mécanisme génétique est présent dans plus de 90 % des cas de sarcome d’Ewing, ce qui en fait une caractéristique diagnostique majeure.
Prédispositions génétiques
Bien que la translocation génétique soit acquise plutôt qu’héritée, certaines prédispositions génétiques peuvent augmenter le risque de développer un sarcome d’Ewing. Des mutations dans des gènes liés à la réparation de l’ADN ou à la stabilité génomique, bien que rares, peuvent contribuer à cette susceptibilité. Cependant, il est important de noter qu’aucune mutation familiale spécifique n’a été directement liée au sarcome d’Ewing, contrairement à d’autres cancers comme le rétinoblastome ou le syndrome de Li-Fraumeni.
Influence de l’âge et du développement
Le sarcome d’Ewing survient le plus souvent pendant l’adolescence, une période marquée par une croissance rapide des os. Cette observation suggère que des facteurs liés au développement, comme l’activité accrue des cellules ostéogéniques (productrices d’os), pourraient jouer un rôle dans l’émergence de la maladie. Ces cellules en prolifération active pourraient être plus vulnérables aux altérations génétiques ou à des perturbations du microenvironnement osseux.
Facteurs environnementaux et immunitaires
Contrairement à de nombreux autres cancers, le sarcome d’Ewing ne semble pas être directement associé à des expositions environnementales telles que les toxines ou les radiations. Toutefois, le rôle du système immunitaire dans la surveillance tumorale est un sujet d’intérêt. Une faiblesse de la réponse immunitaire innée ou adaptative pourrait permettre à des cellules présentant des anomalies génétiques de proliférer sans être éliminées.
Absence de facteurs de risque comportementaux
Il est important de souligner qu’aucun lien n’a été établi entre le sarcome d’Ewing et des facteurs de risque modifiables tels que le tabagisme, l’alimentation ou le mode de vie. Cela distingue cette maladie des cancers de l’adulte souvent influencés par ces variables.
Symptômes du sarcome d’Ewing
Le sarcome d’Ewing est une tumeur osseuse maligne rare qui se manifeste principalement chez les enfants et les adolescents. Ses symptômes varient selon la localisation de la tumeur, son stade et son impact sur les tissus environnants. Ces manifestations cliniques, souvent insidieuses au début, peuvent être confondues avec d’autres conditions musculo-squelettiques, retardant ainsi le diagnostic. Voici une description détaillée des principaux symptômes associés à cette pathologie.
Douleur osseuse localisée
Le symptôme le plus courant du sarcome d’Ewing est une douleur osseuse persistante et localisée. Cette douleur, souvent décrite comme sourde ou lancinante, s’aggrave généralement avec le temps. Elle est initialement intermittente mais devient plus constante à mesure que la tumeur progresse. Contrairement aux douleurs liées à des traumatismes ou à des efforts physiques, celle du sarcome d’Ewing ne disparaît pas avec le repos et peut même s’intensifier la nuit.
Gonflement et masse palpable
Dans les zones où la tumeur est proche de la surface du corps, comme les membres, un gonflement ou une masse palpable peut être observé. Ce gonflement est souvent associé à une chaleur locale et à une rougeur de la peau, en raison de l’inflammation et de la croissance tumorale. La masse peut être douloureuse au toucher, ce qui distingue le sarcome d’autres conditions comme les kystes osseux.
Diminution de la fonction et raideur
La localisation de la tumeur influence directement la mobilité et la fonctionnalité de la zone affectée. Par exemple, si le sarcome d’Ewing se développe dans les membres, les patients peuvent ressentir une limitation de leurs mouvements, une faiblesse musculaire ou une raideur articulaire. Ces symptômes peuvent également affecter la posture ou la démarche, en particulier lorsque la tumeur touche des os porteurs comme le fémur ou le bassin.
Symptômes systémiques
Au fur et à mesure que la maladie progresse, des symptômes systémiques peuvent apparaître, reflétant l’impact général du cancer sur l’organisme. Parmi ces symptômes figurent :
- Fièvre inexpliquée : souvent légère mais persistante, elle peut être confondue avec une infection.
- Fatigue intense : une sensation de faiblesse généralisée, due à la réponse inflammatoire ou à l’anémie causée par la maladie.
- Perte de poids non intentionnelle : fréquemment observée dans les cancers avancés, elle reflète une altération du métabolisme.
Fractures pathologiques
Dans certains cas, la tumeur affaiblit la structure osseuse, augmentant ainsi le risque de fractures dites pathologiques. Ces fractures se produisent sans traumatisme significatif ou lors d’activités quotidiennes banales. Elles constituent souvent un signe révélateur, conduisant à des examens plus approfondis.
Symptômes en fonction de la localisation
La localisation de la tumeur influence grandement les symptômes spécifiques :
- Colonne vertébrale : compression des nerfs ou de la moelle épinière, entraînant des douleurs irradiantes, une perte de sensation ou une paralysie partielle.
- Bassin : douleurs au bas du dos ou dans la région pelvienne, parfois confondues avec des troubles musculo-squelettiques ou gynécologiques.
- Côtes : douleurs thoraciques et difficultés respiratoires, surtout si la tumeur affecte la cage thoracique.
Symptômes tardifs
Lorsque le sarcome d’Ewing est diagnostiqué tardivement, des symptômes dus aux métastases peuvent survenir. Les poumons, les os et la moelle osseuse étant les sites les plus fréquents de propagation, les patients peuvent présenter une toux persistante, des douleurs osseuses diffuses ou une anémie sévère.
Diagnostics différentiels du sarcome d’Ewing
- Ostéomyélite : Une infection osseuse peut présenter des symptômes similaires, tels que douleur osseuse et fièvre. Les radiographies peuvent montrer des lésions lytiques, mais les résultats des tests sanguins, comme la numération globulaire, peuvent aider à différencier l’ostéomyélite du sarcome d’Ewing.
- Chondrosarcome : Un chondrosarcome est un type de cancer du cartilage qui peut affecter les os. Il peut parfois être difficile de distinguer radiologiquement le chondrosarcome du sarcome d’Ewing en raison de similitudes dans les lésions et les réactions périostées.
- Ostéosarcome : Un ostéosarcome est un autre type de cancer osseux qui peut être confondu avec le sarcome d’Ewing en raison de la présence de lésions osseuses destructrices. Cependant, l’emplacement des lésions et les caractéristiques histologiques peuvent aider à différencier les deux.
- Fibrome non ossifiant (lésion fibreuse corticale) : Cette affection bénigne peut provoquer des lésions lytiques semblables à celles observées dans le sarcome d’Ewing. Les caractéristiques radiologiques, ainsi que l’absence de symptômes systémiques, peuvent orienter le diagnostic.
- Granulome éosinophile : Cette lésion bénigne peut être confondue avec le sarcome d’Ewing en raison de ses caractéristiques radiologiques, mais elle ne présente généralement pas la même agressivité et n’a pas de potentiel malin.
- Tumeurs neurogènes malignes : Certaines tumeurs neurogènes malignes, telles que le neuroblastome ou le rhabdomyosarcome, peuvent également être considérées dans les diagnostics différentiels, en particulier lorsqu’elles se produisent dans les mêmes zones anatomiques.
Diagnostic
Méthodes d’Imagerie
Les méthodes d’imagerie utilisées pour diagnostiquer le sarcome d’Ewing incluent :
- Radiographies (RX) : Utilisées pour détecter des anomalies osseuses telles que des lésions lytiques et des réactions périostées.
- IRM (Imagerie par Résonance Magnétique) : Fournit des images détaillées des tissus mous et de l’os pour évaluer la taille de la tumeur, son extension dans les tissus environnants, et la présence de métastases.
- Scintigraphie osseuse : Utilisée pour détecter les zones de lésions osseuses actives ou anormales, ce qui peut aider à évaluer la propagation du cancer.
Biopsie et Confirmation Histologique
- Biopsie : Prélèvement d’un échantillon de tissu de la tumeur pour examen microscopique. La biopsie peut être effectuée par une aiguille fine (biopsie à l’aiguille) ou par une intervention chirurgicale (biopsie chirurgicale).
- Confirmation Histologique : Les échantillons de biopsie sont examinés par un pathologiste pour identifier les caractéristiques microscopiques du sarcome d’Ewing. Cela inclut la présence de cellules tumorales rondes et l’identification de la translocation chromosomique caractéristique (t(11;22)(q24;q12)) par techniques telles que la PCR (réaction en chaîne par polymérase).
Traitement
Approches Thérapeutiques Actuelles
Les principales approches thérapeutiques utilisées pour traiter le sarcome d’Ewing comprennent :
- Chirurgie : La chirurgie est souvent utilisée pour enlever la tumeur primaire, ainsi que pour traiter les métastases localisées dans les os ou les tissus mous.
- Chimiothérapie : Une combinaison de médicaments anticancéreux est administrée pour réduire la taille de la tumeur avant la chirurgie (néoadjuvante) ou après pour éliminer les cellules cancéreuses résiduelles (adjuvante).
- Radiothérapie : Utilisée pour détruire les cellules cancéreuses à l’aide de rayonnements ionisants, souvent en complément de la chirurgie ou de la chimiothérapie.
Nouvelles Thérapies et Recherches en Cours
Des avancées sont en cours pour développer de nouvelles approches thérapeutiques pour le sarcome d’Ewing, telles que :
- Thérapies ciblées : Visent spécifiquement les anomalies génétiques responsables du sarcome d’Ewing, comme les inhibiteurs de tyrosine kinase.
- Immunothérapie : Stimule le système immunitaire à cibler et détruire les cellules cancéreuses, y compris les thérapies basées sur les cellules CAR-T.
- Thérapies géniques : Modifient génétiquement les cellules pour cibler et attaquer les cellules cancéreuses de manière spécifique.
Ces nouvelles approches visent à améliorer l’efficacité du traitement, réduire les effets secondaires et améliorer la qualité de vie des patients atteints de sarcome d’Ewing.
Pronostique et Survie
Facteurs Pronostiques
Les facteurs qui influencent le pronostic du sarcome d’Ewing comprennent :
- Âge au moment du diagnostic : Les enfants plus jeunes ont généralement de meilleures perspectives de guérison.
- Localisation de la tumeur : Les sarcomes d’Ewing localisés dans des zones comme les membres ont un meilleur pronostic que ceux dans des sites plus complexes ou métastatiques.
- Taille et stade de la tumeur : Les tumeurs plus petites et confinées ont un pronostic plus favorable que les tumeurs plus grandes ou métastatiques.
- Réponse au traitement initial : Une réponse positive à la chimiothérapie et à d’autres traitements peut améliorer les perspectives de survie.
- Présence de métastases : Les tumeurs métastatiques ont un pronostic plus réservé que celles qui sont localisées.
Survie à Long Terme et Gestion des Séquelles
- Taux de survie global : Le taux de survie à cinq ans pour le sarcome d’Ewing varie en fonction de plusieurs facteurs, mais globalement, il est d’environ 50 à 70 % pour les cas non métastatiques et d’environ 15 à 30 % pour les cas métastatiques.
- Gestion des Séquelles : Les survivants de sarcome d’Ewing peuvent faire face à des défis à long terme, y compris des problèmes de mobilité, des effets secondaires du traitement, et des préoccupations psychosociales. Une surveillance régulière et un soutien multidisciplinaire sont essentiels pour gérer ces séquelles.
Signes Radiographies du sarcome d’Ewing
Le sarcome d’Ewing est une tumeur osseuse maligne qui se manifeste par des caractéristiques radiographiques distinctives. Les images radiologiques jouent un rôle crucial dans le diagnostic, l’évaluation de l’étendue de la maladie et la planification du traitement. Ces signes varient en fonction du stade de la tumeur, de sa localisation, et de son impact sur les structures osseuses et les tissus environnants. Voici une description détaillée des principaux signes radiographiques associés au sarcome d’Ewing.
1. Destruction osseuse
L’un des signes radiographiques caractéristiques du sarcome d’Ewing est une destruction osseuse agressive. Celle-ci peut se présenter sous différentes formes, reflétant la progression de la maladie :
- Lytique : Des zones de destruction osseuse apparaissent sous forme de lésions claires et irrégulières, indiquant une résorption active de l’os.
- Mixte : Une combinaison de zones lytique et scléreuse peut être observée, traduisant une alternance entre destruction osseuse et tentative de réparation.
- Miteuse : Cette apparence, comparable à des trous d’aspect mité, témoigne d’une destruction rapide et diffuse, fréquente dans le sarcome d’Ewing.
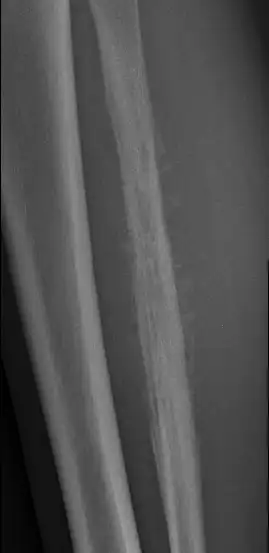
2. Réaction périostée
Une réaction périostée intense est un autre signe typique du sarcome d’Ewing. La tumeur, en envahissant la couche externe de l’os (le périoste), provoque une réponse de ce dernier, se manifestant par plusieurs types de schémas radiographiques :
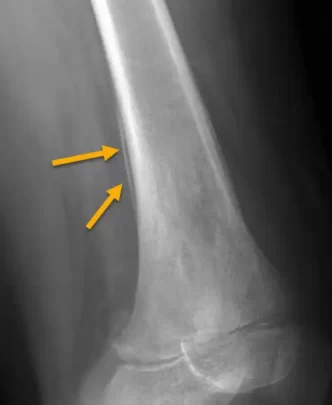
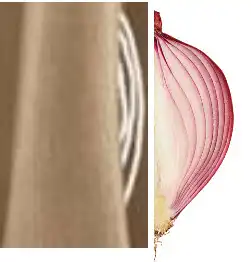
- Aspect en « pelure d’oignon » : Une des caractéristiques les plus spécifiques, cette réaction périostée en couches successives est le résultat de la séparation progressive des couches périostées par la croissance tumorale.
- Aspect en « rayons de soleil » : Dans certains cas, la tumeur entraîne une formation osseuse en éventail, avec des extensions radiales à partir de l’os affecté.
- Réaction périostée spiculée : Des projections osseuses minces et irrégulières s’étendent à partir de l’os, reflétant une réaction rapide et agressive.
3. Extension des tissus mous
Le sarcome d’Ewing est souvent associé à une extension importante vers les tissus mous environnants, visible sur les radiographies avancées ou d’autres imageries comme l’IRM. Bien que l’extension elle-même soit mieux visualisée par des techniques complémentaires, elle peut être suspectée sur les radiographies standard par :
- Une perte de contour clair entre l’os et les tissus adjacents.
- Une augmentation de la densité dans les tissus mous adjacents à la lésion osseuse.
4. Localisation fréquente et distribution
Le sarcome d’Ewing affecte principalement les os longs des membres inférieurs, le bassin et, dans certains cas, les os plats comme les côtes. Les radiographies révèlent des anomalies spécifiques selon la localisation :
- Fémur ou tibia : Des lésions sont généralement observées dans la métaphyse ou la diaphyse, avec des signes d’invasion agressive.
- Bassin : Les radiographies peuvent montrer des lésions destructrices étendues, souvent associées à une masse importante.
- Côtes : Des irrégularités osseuses et des érosions sont visibles, souvent accompagnées d’un élargissement.
5. Fractures pathologiques
En raison de la destruction osseuse importante, le sarcome d’Ewing peut entraîner des fractures pathologiques, souvent visibles sur les radiographies. Ces fractures se produisent sans traumatisme significatif, reflétant la fragilité de l’os affecté.
6. Imagerie complémentaire
Bien que les radiographies fournissent des informations essentielles, elles ne suffisent pas toujours pour établir un diagnostic définitif. Des techniques d’imagerie complémentaires sont souvent utilisées pour confirmer et préciser les signes observés :
- IRM : Utile pour visualiser l’extension des tissus mous et les limites de la tumeur.
- Scanner (CT) : Fournit une meilleure évaluation des détails osseux.
- Scintigraphie osseuse : Permet de détecter les lésions osseuses multiples.
- PET scan : Identifie les métastases et l’activité métabolique de la tumeur.
7. Signes différentiels
Certains signes radiographiques du sarcome d’Ewing peuvent ressembler à d’autres pathologies, comme l’ostéomyélite, les métastases osseuses ou les tumeurs bénignes (ex. : kystes osseux). Les caractéristiques distinctives, telles que la réaction périostée en pelure d’oignon et la destruction agressive, aident à différencier le sarcome d’Ewing de ces autres conditions.
Conclusion
Le sarcome d’Ewing représente bien plus qu’un défi médical; il incarne une bataille entre la fragilité humaine et une résilience extraordinaire. Les progrès scientifiques, qu’ils concernent la compréhension des mécanismes génétiques, l’imagerie diagnostique ou les traitements novateurs, nous rapprochent chaque jour un peu plus de solutions efficaces et ciblées. Pourtant, ce cancer reste une épreuve poignante, particulièrement pour les enfants et leurs familles.
À travers les ténèbres de cette maladie, l’histoire des survivants et la détermination des chercheurs résonnent comme un rappel puissant que l’espoir persiste, même dans les moments les plus difficiles. Ce blog vise à informer et à sensibiliser sur le sarcome d’Ewing, afin que chaque diagnostic s’accompagne d’une prise en charge plus rapide, d’un soutien renforcé et d’une chance accrue de guérison.
En fin de compte, face à ce type de cancer, le véritable message est celui de la persévérance : celle des enfants, des familles, des soignants et des chercheurs qui, ensemble, prouvent que même les défis les plus redoutables peuvent être surmontés. Que l’image d’un enfant courageux, affrontant l’adversité avec espoir, continue de nous inspirer à repousser les limites de la médecine et à tendre vers un avenir où chaque flamme d’espoir brillera pleinement.
Références
- Grier, H. E. (2010). The Ewing family of tumors. Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatric Clinics of North America, 57(1), 119-129. doi:10.1016/j.pcl.2009.11.006
- Gaspar, N., Hawkins, D. S., Dirksen, U., Lewis, I. J., Ferrari, S., Le Deley, M. C., … & Bernstein, M. L. (2015). Ewing sarcoma: Current management and future approaches through collaboration. Journal of Clinical Oncology, 33(27), 3036-3046. doi:10.1200/JCO.2014.59.5256
- Leavey, P. J., Mascarenhas, L., & Marina, N. (2008). Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children’s Oncology Group. Pediatric Blood & Cancer, 51(3), 334-338. doi:10.1002/pbc.21453
- Balamuth, N. J., & Womer, R. B. (2010). Ewing’s sarcoma. The Lancet Oncology, 11(2), 184-192. doi:10.1016/S1470-2045(09)70286-2
- Grünewald, T. G., & Cidre-Aranaz, F. (2020). Ewing sarcoma. Cold Spring Harbor Perspectives in Medicine, 10(4), a037258. doi:10.1101/cshperspect.a037258
- Delattre, O., Zucman, J., Plougastel, B., Desmaze, C., Melot, T., Peter, M., … & Thomas, G. (1992). Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature, 359(6391), 162-165. doi:10.1038/359162a0
- Bernstein, M., Kovar, H., Paulussen, M., Randall, R. L., Schuck, A., Teot, L. A., … & SDCRG (European Intergroup) and POETIC (2016). Ewing’s sarcoma family of tumors: Current management. The Oncologist, 21(5), 576-583. doi:10.1634/theoncologist.2015-0485
- Cotterill, S. J., Ahrens, S., Paulussen, M., Jürgens, H. F., & Voûte, P. A. (2000). Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. Journal of Clinical Oncology, 18(17), 3108-3114. doi:10.1200/JCO.2000.18.17.3108
- Ladenstein, R., Pötschger, U., Le Deley, M. C., Whelan, J., Paulussen, M., Oberlin, O., … & Koscielniak, E. (2010). Primary disseminated multifocal Ewing sarcoma: Results of the Euro-EWING 99 trial. Journal of Clinical Oncology, 28(20), 3284-3291. doi:10.1200/JCO.2009.26.7258
- Stahl, M., Ranft, A., Paulussen, M., Bölling, T., Vieth, V., Bielack, S., & Görtitz, I. (2008). Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatric Blood & Cancer, 51(3), 364-371. doi:10.1002/pbc.21503
- DuBois, S. G., & Krailo, M. D. (2012). Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: A report from the Children’s Oncology Group. Cancer, 118(22), 5477-5483. doi:10.1002/cncr.27597
- Raney, R. B., Asmar, L., Newton, W. A., Bagwell, C., Breneman, J. C., Crist, W. M., … & Wharam, M. D. (1998). Ewing’s sarcoma of soft tissues in childhood: A report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. Journal of Clinical Oncology, 16(12), 3706-3711. doi:10.1200/JCO.1998.16.12.3706
- Bacci, G., Ferrari, S., Longhi, A., Forni, C., Bacchini, P., Donati, D., … & Mercuri, M. (2006). High-dose chemotherapy and autologous haematopoietic rescue in very high-risk metastatic Ewing’s sarcoma. Bone Marrow Transplantation, 37(6), 553-559. doi:10.1038/sj.bmt.1705292
- Casey, D. L., & Wexler, L. H. (2013). The continuing challenge of tumor heterogeneity in Ewing sarcoma. Pediatric Blood & Cancer, 60(6), 865-866. doi:10.1002/pbc.24533
- Hsieh, M. S., Lu, M. Y., Chiang, W. C., Chang, Y. L., Jou, S. T., Lin, D. T., … & Lin, K. H. (2011). The role of chemotherapy for localized Ewing’s sarcoma in children and adolescents: Comparison between Asia and Europe. Journal of Pediatric Hematology/Oncology, 33(6), 423-428. doi:10.1097/MPH.0b013e31821d3369




")












{kind=link}