

tunesharemore_vert
The image of a child in pain tugs at my heart, but it also radiates a message of hope and unwavering resilience. It captures the essence of perseverance and courage in the face of adversity. It reminds me that even in the deepest darkness, hope still flickers, like an indomitable flame.
Introduction
Ewing sarcoma is a tumor that forms in the bones or soft tissues. This type of cancer occurs mainly in children and young adults, often appearing during adolescence. Ewing sarcomas are the second most common primary malignant bone tumors of childhood after osteosarcoma. They usually arise from the medullary cavity with invasion of the Haversian system. This sarcoma was first described by James Ewing, a pioneer in the field of cancer research, in 1921.
Ewing sarcoma mainly affects the long bones, including the femur, tibia, and humerus. It can spread (metastasize) to other parts of the body. Although relatively rare, about 1.7 children under the age of 15 will develop this cancer.

Ewing sarcoma can spread (metastasize) to other parts of the body. This cancer is relatively rare. About 1.7 million children under the age of 15 will have this cancer.
Understanding Ewing Sarcoma: How it Forms
Ewing sarcoma is a cancer that develops in the bones or soft tissues, mainly in young people. Here’s how it forms:
The Key: Genetic Anomaly
- Chromosomal Mix-Up : Ewing sarcoma starts with a genetic problem called a translocation. This means that parts of chromosomes 11 and 22 mix incorrectly, creating a bizarre new gene called EWSR1-FLI1.
- Effects of this New Gene : This bizarre gene produces an abnormal protein. This protein disrupts the normal growth of bone cells, preventing them from properly differentiating into specialized cells. This leads to out-of-control cell growth, a hallmark of cancer.
- Impact on the Body : The abnormal protein activates signals in cells that promote rapid tumor growth. It also encourages the formation of new blood vessels around the tumor, ensuring its supply of nutrients and oxygen.
In short, Ewing’s sarcoma is caused by a genetic problem that disrupts normal bone growth, leading to the formation of a malignant tumor.
Causes of Ewing’s Sarcoma
The exact causes of Ewing sarcoma are not completely understood, but it is widely accepted that this tumor results from specific genetic mutations. Ewing sarcoma is associated with a characteristic chromosomal abnormality, primarily a t(11;22)(q24;q12) translocation, involving the genes EWSR1 (Ewing Sarcoma breakpoint region 1) on chromosome 22 and FLI1 (Friend Leukemia Integration 1) on chromosome 11. This translocation generates a novel EWSR1-FLI1 fusion gene, which plays a key role in tumor development.
Other genetic factors and predispositions may also contribute to the development of Ewing sarcoma, including:
- Age: Ewing sarcoma is most common in children and young adults, usually in their teens to 20s. However, it can also occur in older adults.
- Race: There is a slight prevalence of Ewing sarcoma in Caucasian individuals.
- Gender: Ewing sarcoma has been observed to affect men slightly more frequently than women.
- Family history: Although Ewing sarcoma is not generally considered to be heritable, a family history of bone cancers may increase the risk in some individuals.
- Radiation exposure: Previous exposure to radiation, although rare, has been identified as a potential risk factor for the development of Ewing sarcoma.
Symptoms of Ewing Sarcoma
- Bone pain: Pain in the affected area is one of the most common symptoms of Ewing sarcoma. The pain may be persistent and gradually increase over time.
- Swelling: A swelling or swelling in the affected area may be observed. This may be due to the growth of the tumor and the reaction of the surrounding tissues.
- Joint stiffness: If the tumor affects a joint, joint stiffness may develop. This can lead to decreased mobility in the affected area.
- Fever: Some patients may experience fever, although this is not a symptom specific to Ewing sarcoma. Fever may indicate a systemic inflammatory reaction.
- Fatigue: Persistent fatigue may accompany Ewing sarcoma, often associated with the presence of a malignant tumor and the immune system’s response.
- Weight loss: Unexplained weight loss may occur, primarily due to systemic effects of the disease.
- Spontaneous fractures: In some cases, the tumor can weaken the bone to the point of causing spontaneous fractures or cracks.
- Neurological symptoms: If the tumor compresses nerves or the spinal cord, neurological symptoms such as numbness, tingling, or weakness may occur.
Differential diagnoses of Ewing sarcoma
- Osteomyelitis: A bone infection can have similar symptoms, such as bone pain and fever. X-rays may show lytic lesions, but blood test results, such as blood counts, can help differentiate osteomyelitis from Ewing sarcoma.
- Chondrosarcoma: Chondrosarcoma is a type of cartilage cancer that can affect bones. It can sometimes be difficult to distinguish chondrosarcoma from Ewing sarcoma radiologically because of similarities in the lesions and periosteal reactions.
- Osteosarcoma: Osteosarcoma is another type of bone cancer that can be confused with Ewing sarcoma due to the presence of destructive bone lesions. However, the location of the lesions and histologic features can help differentiate the two.
- Nonossifying fibroma (cortical fibrous lesion): This benign condition may cause lytic lesions similar to those seen in Ewing sarcoma. Radiological features, as well as the absence of systemic symptoms, can guide the diagnosis.
- Eosinophilic granuloma: This benign lesion can be confused with Ewing’s sarcoma due to its radiological characteristics, but it generally does not present the same aggressiveness and does not have malignant potential.
- Malignant neurogenic tumors: Some malignant neurogenic tumors, such as neuroblastoma or rhabdomyosarcoma, may also be considered in the differential diagnoses, especially when they occur in the same anatomic areas.
Diagnostic
Imaging Methods
Imaging methods used to diagnose Ewing sarcoma include:
- Radiographs (RX) : Used to detect bone abnormalities such as lytic lesions and periosteal reactions.
- MRI (Magnetic Resonance Imaging) : Provides detailed images of soft tissues and bone to assess tumor size, its spread into surrounding tissues, and the presence of metastases.
- Bone scan : Used to detect areas of active or abnormal bone lesions, which can help assess the spread of cancer.
Biopsy and Histological Confirmation
- Biopsy : Taking a sample of tissue from the tumor for microscopic examination. The biopsy may be performed with a fine needle (needle biopsy) or by surgery (surgical biopsy).
- Histologic Confirmation : Biopsy specimens are examined by a pathologist to identify microscopic features of Ewing sarcoma. This includes the presence of round tumor cells and identification of the characteristic chromosomal translocation (t(11;22)(q24;q12)) by techniques such as PCR (polymerase chain reaction).
Treatment
Current Therapeutic Approaches
The main therapeutic approaches used to treat Ewing sarcoma include:
- Surgery : Surgery is often used to remove the primary tumor, as well as to treat metastases located in bone or soft tissues.
- Chemotherapy : A combination of anticancer drugs is given to shrink the tumor before surgery (neoadjuvant) or afterward to eliminate residual cancer cells (adjuvant).
- Radiotherapy : Used to destroy cancer cells using ionizing radiation, often in addition to surgery or chemotherapy.
New Therapies and Current Research
Advances are underway to develop new therapeutic approaches for Ewing sarcoma, such as:
- Targeted therapies : Specifically target the genetic abnormalities responsible for Ewing sarcoma, such as tyrosine kinase inhibitors.
- Immunotherapy : Stimulates the immune system to target and destroy cancer cells, including CAR-T cell-based therapies.
- Gene therapies : Genetically modify cells to specifically target and attack cancer cells.
These new approaches aim to improve treatment effectiveness, reduce side effects and improve the quality of life of patients with Ewing sarcoma.
Prognosis and Survival
Prognostic Factors
Factors that influence the prognosis of Ewing sarcoma include:
- Age at diagnosis : Younger children generally have a better outlook for recovery.
- Tumor location : Ewing sarcomas located in areas such as the limbs have a better prognosis than those in more complex or metastatic sites.
- Tumor size and stage : Smaller, confined tumors have a more favorable prognosis than larger or metastatic tumors.
- Response to initial treatment : A positive response to chemotherapy and other treatments may improve survival prospects.
- Presence of metastases : Metastatic tumors have a more reserved prognosis than those that are localized.
Long-Term Survival and Management of Sequelae
- Overall survival rate : The five-year survival rate for Ewing sarcoma varies depending on several factors, but overall it is about 50 to 70 percent for nonmetastatic cases and about 15 to 30 percent for metastatic cases.
- Management of Sequelae : Survivors of Ewing sarcoma may face long-term challenges, including mobility problems, treatment side effects, and psychosocial concerns. Regular monitoring and multidisciplinary support are essential to manage these sequelae.
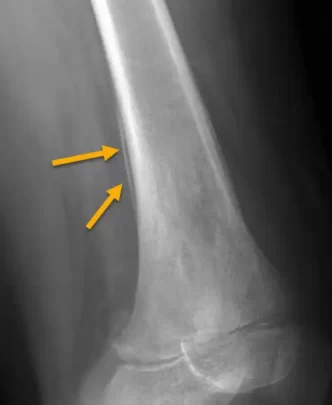
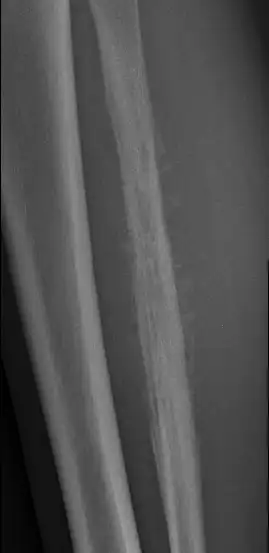
Signs of Ewing’s Sarcoma X-rays
- Lytic lesions: On X-rays, Ewing sarcoma may appear as areas of destroyed bone, with lytic lesions. These areas appear dark and irregular, indicating destruction of bone tissue.
- Soft tissue infiltration: Ewing sarcoma may spread to surrounding soft tissues. On radiographs, this may be seen as increased soft tissue density adjacent to the affected bone.
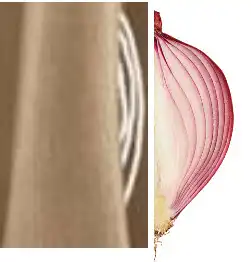
- Periosteal reaction: A periosteal reaction, characterized by the formation of new bone in the periosteum (the outer membrane of the bones), may be visible on X-rays. This may give the bone an “onion” appearance or cause thickening of the periosteum.
- Mass formation: In advanced cases, bone mass formation may be visible on X-rays. This mass can disrupt the normal structure of the bone and surrounding tissues.
- Pervasive destruction: Radiographs of Ewing sarcoma may reveal widespread and pervasive destruction of the affected bone. This feature contributes to understanding the aggressiveness of the tumor in its ability to invade bone tissue.
- Aggressive periosteal reaction: In addition to the periosteal reaction mentioned above, Ewing sarcoma may present with a particularly aggressive periosteal reaction, marked by thickening and exacerbated osteoblastic activity.
- Onion skin appearance: The onion skin appearance refers to the layered or ring-like arrangement of lytic and sclerosing areas on radiographs. This appearance may be the result of rapid tumor growth and alternation between areas of destruction and areas where new bone is formed.
These radiographic features, combined with other clinical and diagnostic elements, contribute to establishing the diagnosis of Ewing sarcoma. It is crucial to note that these signs are not specific to Ewing sarcoma and can also be observed in other bone pathologies. A definitive diagnosis requires a multidisciplinary approach with the participation of specialists in oncology, radiology and pathology.
References
- Grier, H. E. (2010). The Ewing family of tumors. Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatric Clinics of North America, 57(1), 119-129. doi:10.1016/j.pcl.2009.11.006
- Gaspar, N., Hawkins, D. S., Dirksen, U., Lewis, I. J., Ferrari, S., Le Deley, M. C., … & Bernstein, M. L. (2015). Ewing sarcoma: Current management and future approaches through collaboration. Journal of Clinical Oncology, 33(27), 3036-3046. doi:10.1200/JCO.2014.59.5256
- Leavey, P. J., Mascarenhas, L., & Marina, N. (2008). Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children’s Oncology Group. Pediatric Blood & Cancer, 51(3), 334-338. doi:10.1002/pbc.21453
- Balamuth, N. J., & Womer, R. B. (2010). Ewing’s sarcoma. The Lancet Oncology, 11(2), 184-192. doi:10.1016/S1470-2045(09)70286-2
- Grünewald, T. G., & Cidre-Aranaz, F. (2020). Ewing sarcoma. Cold Spring Harbor Perspectives in Medicine, 10(4), a037258. doi:10.1101/cshperspect.a037258
- Delattre, O., Zucman, J., Plougastel, B., Desmaze, C., Melot, T., Peter, M., … & Thomas, G. (1992). Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature, 359(6391), 162-165. doi:10.1038/359162a0
- Bernstein, M., Kovar, H., Paulussen, M., Randall, R. L., Schuck, A., Teot, L. A., … & SDCRG (European Intergroup) and POETIC (2016). Ewing’s sarcoma family of tumors: Current management. The Oncologist, 21(5), 576-583. doi:10.1634/theoncologist.2015-0485
- Cotterill, S. J., Ahrens, S., Paulussen, M., Jürgens, H. F., & Voûte, P. A. (2000). Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. Journal of Clinical Oncology, 18(17), 3108-3114. doi:10.1200/JCO.2000.18.17.3108
- Ladenstein, R., Pötschger, U., Le Deley, M. C., Whelan, J., Paulussen, M., Oberlin, O., … & Koscielniak, E. (2010). Primary disseminated multifocal Ewing sarcoma: Results of the Euro-EWING 99 trial. Journal of Clinical Oncology, 28(20), 3284-3291. doi:10.1200/JCO.2009.26.7258
- Stahl, M., Ranft, A., Paulussen, M., Bölling, T., Vieth, V., Bielack, S., & Görtitz, I. (2008). Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatric Blood & Cancer, 51(3), 364-371. doi:10.1002/pbc.21503
- DuBois, S. G., & Krailo, M. D. (2012). Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: A report from the Children’s Oncology Group. Cancer, 118(22), 5477-5483. doi:10.1002/cncr.27597
- Raney, R. B., Asmar, L., Newton, W. A., Bagwell, C., Breneman, J. C., Crist, W. M., … & Wharam, M. D. (1998). Ewing’s sarcoma of soft tissues in childhood: A report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. Journal of Clinical Oncology, 16(12), 3706-3711. doi:10.1200/JCO.1998.16.12.3706
- Bacci, G., Ferrari, S., Longhi, A., Forni, C., Bacchini, P., Donati, D., … & Mercuri, M. (2006). High-dose chemotherapy and autologous haematopoietic rescue in very high-risk metastatic Ewing’s sarcoma. Bone Marrow Transplantation, 37(6), 553-559. doi:10.1038/sj.bmt.1705292
- Casey, D. L., & Wexler, L. H. (2013). The continuing challenge of tumor heterogeneity in Ewing sarcoma. Pediatric Blood & Cancer, 60(6), 865-866. doi:10.1002/pbc.24533
- Hsieh, M. S., Lu, M. Y., Chiang, W. C., Chang, Y. L., Jou, S. T., Lin, D. T., … & Lin, K. H. (2011). The role of chemotherapy for localized Ewing’s sarcoma in children and adolescents: Comparison between Asia and Europe. Journal of Pediatric Hematology/Oncology, 33(6), 423-428. doi:10.1097/MPH.0b013e31821d3369

















{kind=link}